
Disorders of N-Glycan Processing

Congenital Disorders of Glycosylation (CDG) are multisystem disorders caused by defects in glycoprotein biosynthesis. Changes in glycosylation are a major feature of CDGs, making Glycan Analysis the most promising method for screening CDGs. CD BioGlyco has been concentrating on glycobiology research for many years and has developed an advanced Glycomics Platform, providing glycan isolation, purification, and characterization services to our clients.
During N-glycosylation, the fully assembled lipid-linked oligosaccharide (LLO) is transferred to the asparagine (Asn) glycosylation site in the nascent protein under the catalysis of oligosaccharyltransferase (OST). Then LLO is processed generally by the glucosidase-dependent pathway in the endoplasmic reticulum (ER). The first step is the removal of the outer two α-1,2-linked glucose (Glc) residues by α-glucosidase I (MOGS) and α-glucosidase II from the transferred N-glycan GlcNAc2Man9Glc3. Binding of the generated GlcNAc2Man9Glc to calnexin (CNX) and calreticulin (CRT) and partial protein refolding. α-Glucosidase II then trims the remaining Glc residues. If the folding of the glycoprotein is incomplete, N-glycan is transiently re-glycosylated to GlcMan9GlcNAc2 by UDP-Glc: glycoprotein glucosyltransferase (UGGT). This process, known as the calnexin cycle, prevents further trimming of N-glycans by glycoproteins that are not yet properly folded. Correctly folded glycoproteins exit the ER while misfolded glycoproteins enter the endoplasmic reticulum-associated degradation (ERAD) pathway.
The first step in ERAD is the trimming of the mannose (Man) of the N-glycan by α-mannosidase to generate the Man7 structure. The Man7 structure is then recognized by ER-resident lectins OS-9 and XTP-3B, which retrogradely translocate misfolded glycoproteins back to the cytoplasm. In the cytosol, the misfolded glycoprotein is de-N-glycosylated by N-glycanase 1 (NGLY1), resulting in free N-glycans and misfolded proteins. Misfolded protein is subsequently degraded by the proteasome, while released N-glycan is further processed in the cytosol and finally fully degraded in the lysosome.
In the cis-Golgi apparatus, Golgi mannosidase I catalyze the removal of all α-1,2-linked Man residues on properly folded N-glycans. N-Acetylglucosamine transferase I (GnT-I, MGAT1) then transfer N-acetylglucosamine (GlcNAc) residues to α1,3 Man to generate GlcNAc2Man5GlcNAc. Golgi α-mannosidase II sequentially removes two α-1,3 linked Man residues. GnT-II (MGAT2) transfers a GlcNAc residue to the newly exposed Man residue, generating a common core structure of biantennary complex glycans GlcNAc2Man3GlcNAc2. In mammals, GnT-III (MGAT3), GnT-IV (MGAT4A and MGAT4B), and GnT-V (MGAT5) catalyze GlcNAc to generate bisected GlcNAc, β1,4-GlcNAc branch and β1,6-GlcNAc branch, respectively. N-Glycans can also be further modified by other types of sugars, including fucose (Fuc), β1,3- or β1,4-galactose (Gal), and α2,3- or α2,6-sialic acid (Sia).
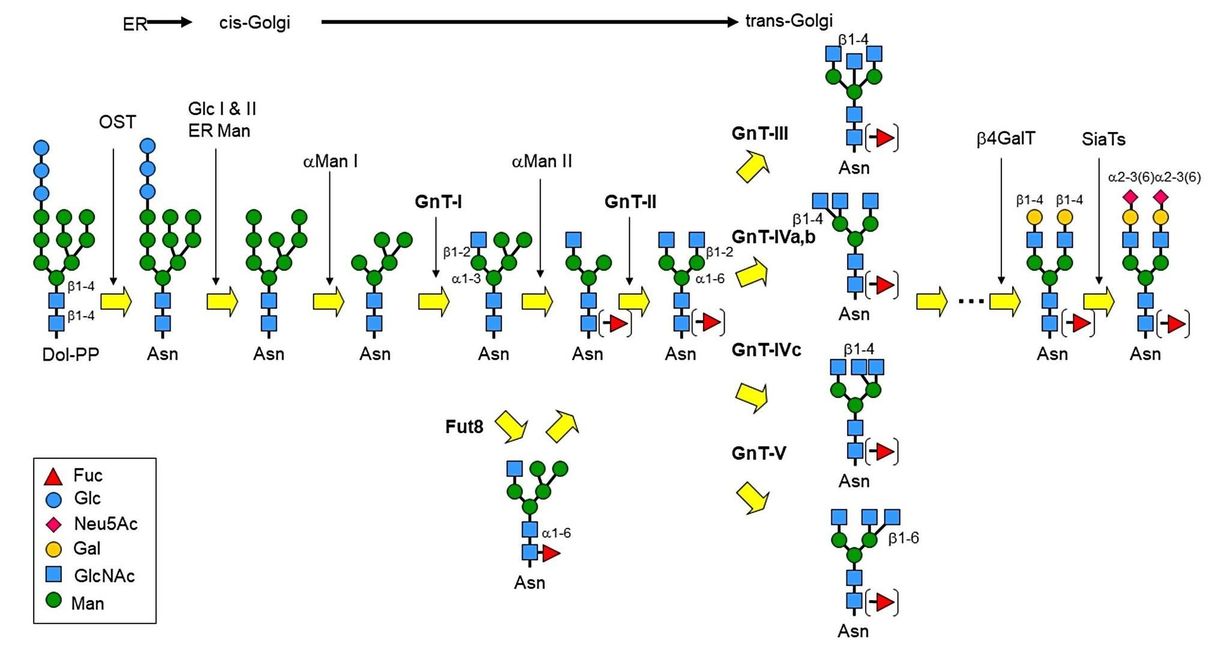 Fig.1 N-Glycan processing. (Nagae, et al., 2020)
Fig.1 N-Glycan processing. (Nagae, et al., 2020)
Aberrant N-Glycosylation leads to specific glycation disorders. Mutations in Golgi glycosyltransferases and transporters have been reported to cause defects in N-glycan processing, resulting in severe CDG. Here, we describe CDGs caused by mutations in several Glycosyltransferases and transporters involved in N-glycan processing.
Defects in MGAT2 cause generalized hypotonia and moderate-to-severe neuromotor developmental delay in CDG-IIa patients in infancy. MGAT2 affects only the Man-α1,6 antennae of glycans, encoded by the MGAT2 gene located on chromosome 14q21.
CDG IIb is a disease caused by mutations in ER glucosidase I. Only one patient from consanguineous marriage has been reported, who had hypotonia, decreased mobility, generalized edema, hypoventilation, and episodes of apnea from birth.
CDG IIc was first discovered in 1992 due to a defect in the GDP-fucose transporter. Patients with CDG IIc are characterized by recurrent bacterial infections in infancy, developmental delay, microcephaly, and moderate psychomotor retardation. The low affinity of fucose transporters may be overcome clinically by increasing fucose levels with oral supplements.
CDG IId is due to the insufficient activity of UDP-Gal: β-GlcNAc β-1,4-galactosyltransferase and glycans exhibit a significant loss of Gal and Sia from transferrin. Patients with CDG IId exhibit moderate psychomotor, delayed physical development, generalized hypotonia, and muscle weakness.
CDG-IIf is a rare genetic disorder that primarily affects the body's vascular system. It is caused by a defect in the CMP-sialic acid transporter encoded by the SLC35A1 gene located on chromosome 6.
CD BioGlyco is dedicated to glycosylation research for many years and provides high-quality Glycoprotein Structural Analysis and Custom Glycosylation Service to meet your research needs. If you are interested in our services, please feel free to contact us for more information.
References:

